
Recibido para arbitraje: 20/07/2017
Aprobado para su publicación: 15/11/2017
El carcinoma oral de células escamosas (COCE) es una neoplasia de gran relevancia internacional en la literatura actual, principalmente por su aumento de incidencia, diagnóstico tardío y baja tasa de supervivencia de los pacientes a los cinco años en comparación con otros tipos de tumores. Siendo un cáncer de fácil acceso al diagnóstico clínico, se enfrenta a limitaciones en la prevención, por la subjetividad del diagnóstico histológico de la displasia epitelial oral y el tratamiento, es importante desarrollar nuevas estrategias para mejorar la situación actual. La transformación de células normales en células cancerosas es un proceso evolutivo en el que las mutaciones somáticas responsables de esta transformación podrían representar objetivos terapéuticos actualmente desconocidos. Por lo tanto, para entender mejor el desarrollo y progresión del COCE, el estudio de base genética es crucial para ampliar el conocimiento de las posibles aberraciones genéticas que pueden estar asociadas con esta condición. Este es un artículo de revisión que propone analizar y discutir los cambios genéticos epiteliales de la mucosa oral durante la etapa de malignización, incorporando nuevas estrategias de investigación con base genética.
Palabras clave: alteraciones genéticas, mucosa oral, carcinoma de células escamosas.
Oral squamous cell carcinoma (OSCC) is a neoplasm with international relevance in the current literature, mainly because of its increased incidence, late diagnosis and low survival rate of patients at five years of age compared to other types of tumors. Being a cancer with an easy access to clinical diagnosis, faces limitations in preventing, by the subjectivity of the histological diagnosis of oral epithelial dysplasia and treatment, it is important to develop new strategies to improve the current situation. The transformation of normal cells to cancerous cells is an evolutionary process, in which somatic mutations responsible for this transformation could represent therapeutic targets currently unknown. Therefore, to better understand the development and progression of OSCC, study of the genetic basis is crucial to expand the knowledge of the possible genetic aberrations that may be associated with this condition. This is a review article that proposes to analyze and discuss the epithelial genetic changes of the oral mucosa during the stage of malignization, incorporating new research strategies based on genetics.
Key words: genetic alterations, oral mucosa, oral squamous cell carcinoma
Autor Correspondiente:
Prof. Ricardo Fernández-Ramires. Dirección: Sergio Livingstone Pohlhammer 943, Independencia, Santiago, Chile. Correo electrónico: [email protected] Teléfono: +56229781718
Los autores declaran no tener conflicto de intereses. Apoyo Financiero: FONDECYT N° 11140281
El carcinoma de células escamosas (CCE) es el más común de todas las neoplasias malignas orales. Se ha observado que entre el 11% y el 36% de leucoplasias con displasia y que hasta el 70% de leucoplasias verrucosas proliferativas pueden convertirse en lesión maligna cada año1, 2.
La etiología del carcinoma oral de células escamosas (COCE) ha sido considerada multifactorial3,4. Específicamente, se diagnostican más de 300.000 casos de cáncer oral por año alrededor del mundo, incluyendo labio, los cuales 50% no llegan con vida a los 5 años. El sur de Brasil, Argentina y Uruguay tienen las mayores tasas de incidencia de cáncer de boca y faringe en América Latina5. Debido a su ubicación, los carcinomas orales y faríngeos también pueden estar asociados con una morbilidad significativa, ya que estos cánceres y tratamientos pueden ser mutiladores y afectar las funciones diarias, lo que impacta negativamente a la calidad de vida. Las tasas de supervivencia se han mantenido relativamente sin cambios durante las últimas tres décadas, probablemente debido al reconocimiento tardío de la enfermedad, como ocurre en la mayoría de los países4. En consecuencia, se hace cada vez más destaque al diagnóstico precoz y monitoreo frecuente de las lesiones precancerosas.
A pesar de estos esfuerzos, los esquemas actuales de clasificación no logran diferenciar con precisión los desórdenes sin potencial progresivo de los con potencial progresivo, siendo así el manejo clínico de los pacientes con estas lesiones no debiera basarse en este criterio exclusivo. Por otro lado, varios estudios han demostrado una gran variabilidad inter e intra-examinador en la evaluación de la presencia o ausencia y el grado de la displasia epitelial oral.6-12 Por todo lo anterior, este trabajo busca revisar qué alteraciones genéticas podrían estar involucradas en el proceso de malignización del epitelio oral, discutiendo la posible utilización de estas variaciones en el diagnóstico personalizado y terapias específicas.
COCE en etapas tempranas a moderadas son frecuentemente tratados quirúrgicamente con radioterapia asociada, además, con o sin administración de quimioterapia en el contexto de adyuvante postoperatorio para pacientes de alto riesgo que tienen ganglios linfáticos comprometidos y/o metástasis que se extienden más allá de la cápsula del ganglio linfático13-14. En la enfermedad avanzada, los enfoques multidisciplinarios no-quirúrgicos se utilizan cada vez con mayor frecuencia para mejorar el control de la enfermedad, prolongar la supervivencia y mantener una calidad de vida aceptable para los pacientes15-17. Aunque se utilice la mejor combinación de abordajes terapéuticos, más del 50% de los pacientes con COCE experimentarán recidiva, ya sea localmente, en los ganglios linfáticos regionales o en un sitio distante14,18. Independientemente de la ubicación o etapa del COCE, se necesitan terapias más efectivas.
La secuenciación del exoma completo de tumores primarios frente a las muestras de metástasis ha sido limitado hasta el momento debido a la falta de muestras, sin embargo, se ha observado que las lesiones metastásicas individuales son de origen clonal y son genéticamente únicas, entretanto, tienen una ascendencia clonal rastreable del tumor primario. Por lo tanto, la diversidad genética subclonal es clave para el éxito o fracaso de la terapia19. Este es un desafío considerable técnico y bioinformático en la genómica del cáncer, que requerirá profunda secuenciación e investigación de los genomas de los patrones de segregación de mutaciones para comprender la diversidad genética dentro de neoplasias y cómo esto cambia en respuesta a las intervenciones.
Una estrategia promisora para el tratamiento del COCE y otros cánceres es la terapia dirigida molecular. En este enfoque, las alteraciones moleculares específicas de una célula cancerosa que contribuyen al fenotipo neoplásico se emplean como dianas de anticuerpos, moléculas pequeñas y/o construcciones genéticas. Este enfoque difiere de las terapias citotóxicas convencionales, que inhiben la actividad mitótica de todas las células en división, dando lugar a efectos tóxicos en todos los tejidos que normalmente tienen una rápida renovación celular, como la mucosa oral, los folículos pilosos y revestimiento intestinal20. Se piensa que la terapia dirigida ofrece un índice terapéutico más alto y por lo tanto se asocia con menos toxicidad que los fármacos citotóxicos. El desarrollo de enfoques dirigidos al COCE requiere comprensión de la patogénesis molecular de la enfermedad, así como una mejor caracterización de los eventos moleculares específicos implicados en el crecimiento, invasión y metástasis del cáncer.
Los desórdenes potencialmente malignos de la mucosa oral (DPMO), incluyen leucoplasia, eritroplasia, liquen plano, fibrosis submucosa oral, lupus eritematoso discoide y queilitis actínica. Las lesiones más frecuentes que preceden al desarrollo del COCE, son leucoplasia y eritroplasia21-24. El diagnóstico clínico de estas lesiones puede ser complejo y se necesita experiencia adecuada para diferenciar clínicamente estas lesiones de otras patologías. Se han sugerido métodos clínicos complementarios21,25-26, pero hasta el momento no se ha determinado un método no invasivo que permita identificar el potencial de riesgo de transformación maligna de una determinada DPMO.
La tasa de transformación maligna de DPMO a COCE varía según la población, sus hábitos y localización de la lesión principalmente 27. El riesgo más alto afecta a los fumadores que han utilizado cigarrillos sin filtrar por muchos años. El alcohol puede actuar como promotor, teniendo un efecto sinérgico con el tabaco28. DPMO en piso de boca y lengua tienen un riesgo aumentado de transformación maligna, así como la edad avanzada27.
La patología clásica enfatiza que las lesiones neoplásicas se caracterizan por anomalías arquitectónicas y citológicas que lo distinguen del tejido normal. Las características citológicas de la neoplasia incluyen agrandamiento nuclear e hipercromatismo, membranas nucleares irregulares, aumento de la proporción núcleo-citoplasma de las células, patrón de cromatina agrupada, nucléolos múltiples, elongación de núcleos, mitosis atípicas y citoplasma hipereosinofílico o denso26,29. Muchas de estas características histopatológicas representan la expresión fenotípica de la pérdida de control de la proliferación y diferenciación celular, como resultado de mutaciones en las vías moleculares que regulan estos aspectos esenciales del crecimiento celular19,29. Sin embargo, paralelamente a los avances recientes, la sincronización y secuencia de eventos moleculares que conducen a la transformación neoplásica, es de nuestro conocimiento de que muchas importantes anomalías moleculares que conducen a la desregulación de la proliferación y diferenciación celular ocurren antes de la expresión morfológica de la neoplasia. Por lo tanto, no todas las lesiones precursoras neoplásicas muestran necesariamente características morfológicas de displasia epitelial (Figura 1). Las alteraciones en el contenido de DNA, el control del ciclo celular y las mutaciones en los genes supresores de tumores, tales como p53 y p16, se producen en una etapa temprana de la transformación neoplásica, antes del desarrollo de características morfológicas convencionales de displasia epitelial.29
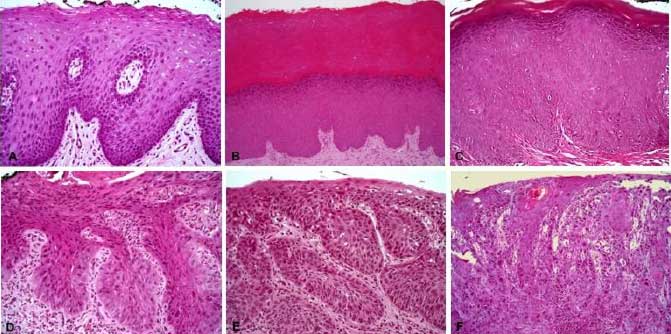
En muchos procesos carcinogénicos, la transformación citológica de las células representa una aberración biológica tardía en la vía carcinogénica que conduce en última instancia a la transformación de una célula normal a maligna. Por lo tanto, con mejorías en nuestra comprensión sobre alteraciones moleculares involucradas en carcinogénesis oral y con la realización de estudios que ayuden a correlacionar genotipo y fenotipo, los patólogos podrán reconocer la presencia de lesiones pre-malignas, que no necesariamente muestran evidencia morfológica de displasia epitelial.29
El cáncer es una enfermedad de evolución dinámica, estocástica somática genómica30-31. Se cree que todos los cánceres surgen como resultado de (i) inestabilidad genómica somática, (ii) generación de diversidad genómica somática, y (iii) selección natural de variantes que subyacen la progresión a cáncer28. Basado principalmente en estudios de cánceres avanzados en un solo punto en el tiempo, se ha inferido que diferentes tipos de inestabilidad genómica tienen diferentes tasas de progresión. Por ejemplo, se cree que el cáncer colorrectal y otros cánceres se desarrollan a través de la acumulación gradual de mutaciones puntuales a una tasa de mutación normal durante décadas32-33, lo que proporcionaría una ventana temporal prolongada de oportunidad para la detección temprana. La carcinogénesis multi-etapa es un proceso evolutivo en el que la inestabilidad genética genera nuevos clones y la selección natural entre tales variantes impulsa expansiones clonales34.
El número de lesiones genéticas necesarias para producir un cáncer es desconocido para la mayoría de los tejidos, pero se ha estimado que es de 2 a 1235. Las tasas normales de mutación en ausencia de expansión clonal no pueden explicar la acumulación de tantas lesiones36-38. Tanto inestabilidad genética y expansiones clonales se observan comúnmente en la progresión neoplásica, y es probable que los dos factores colaboren en la evolución clonal39. Si esto es cierto, entonces la expansión de un clon genéticamente inestable en una lesión premaligna se asociará con el riesgo de progresión al cáncer y, por lo tanto, es posible extraer estos marcadores en el marco del desarrollo de blancos terapéuticos.
La secuenciación del genoma del cáncer, facilitada por la introducción de la segunda generación de secuenciación del genoma completo, ha proporcionado una mejor visión de la complejidad genética y de biología evolutiva de las células cancerosas40. En la mayoría de los casos, la transformación y metástasis son probablemente clonales, porque se derivan de células individuales40. Por lo tanto, la identificación de las mutaciones presentes en todas las células de un tumor puede ayudar a reconstruir el genotipo de la célula precursora. Estos eventos precursores limitan la complejidad genética y clonal de los tumores. Ya tenemos una larga lista de mutaciones recurrentes en muchos tipos de tumores como resultado del mapeo fino de roturas cromosómicas, secuenciación de genes candidatos y selección funcional de muestras a granel de tumores.41
La transformación de células normales en células cancerosas es un proceso evolutivo. Las poblaciones de células precancerosas se reproducen, mutan y compiten por los recursos. Algunas de estas mutaciones conducen eventualmente al cáncer. Aunque cada cáncer se deriva de una sola célula normal, la población de células neoplásicas que constituyen el cáncer final a menudo tiene una historia evolutiva compleja. Se cree que requieren múltiples ondas de expansión clonal, provocadas cada una por una mutación adicional del controlador, para generar el clon dominante que se manifiesta como el cáncer sintomático. A lo largo del camino, los clones de ramificación adicionales con otros controladores pueden haber sido separados por giro que no pudieron superar al clon dominante, el cual por sí mismo pudo haber producido otro clon menor con una mutación adicional del controlador que con el tiempo dominaría. Algunos de estos clones menores pueden haber sido completamente extinguidos, pero otros pueden persistir.42-44
Las mutaciones somáticas adquiridas por células cancerosas a medida que se dividen pueden servir como marcadores de origen clonal y permitir así la reconstrucción retrospectiva del árbol evolutivo de cánceres individuales. Estos análisis han revelado la compleja estructura clonal de ciertos cánceres y han demostrado que los clones menores en la presentación inicial del cáncer son a menudo fuente del clon mayor que se repite después del tratamiento19,42-44. Gran parte del enfoque en investigación genómica del cáncer ha sido comprendido en los genes y vías genéticas de cánceres avanzados para mejorar las estrategias de tratamiento del paciente45-46. Sin embargo, se ha dedicado relativamente poca investigación a la comprensión de la dinámica de la evolución genómica somática que conduce al cáncer y subyace a las observaciones clínicas donde algunas lesiones orales progresan rápidamente al COCE, mientras que el resto no progresa durante toda la vida. (Figura 2)
El uso de muestras de tejido incluido, fijado en formalina e embebido en parafina (FFEP) para análisis de expresión de RNA ha recibido recientemente interés como resultado de técnicas mejoradas para recuperar ácidos nucleicos de muestras de FFEP, así como el gran número de estas muestras a menudo disponibles a los investigadores. El uso de muestras FFEP para el análisis de expresión de RNA requiere superar varios desafíos. La fijación en formalina resulta en la degradación y fragmentación del DNA/RNA, así como la modificación de los nucleótidos, todos los cuales disminuyen el rendimiento de los ácidos nucleicos extraídos de dichas muestras.47-55
Se ha demostrado que la mayoría de los estudios genómicos en muestras FFEP tienen resultados muy similares a las muestras frescas congeladas. Esto es de vital importancia, ya que una extracción de ácidos nucleicos de forma óptima y reproducible en estas muestras nos puede permitir ampliar el estudio genómico de enfermedades específicas cuyos resultados permitirían correlacionar aspectos clínicos, histopatológicos y ser de gran utilidad para tomar decisiones acerca de los posibles tratamientos de estas enfermedades.56-58
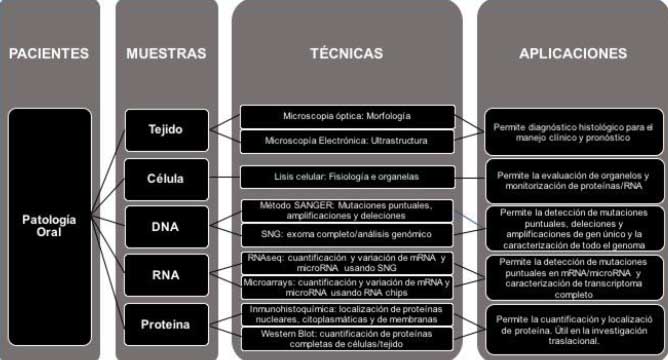
La disección del tejido tumoral y/o pre-maligno y la secuenciación del exoma siguen siendo un área poco explorada en biología oral. La secuenciación de nueva generación (SNG) acelera el proceso de estudio del DNA mediante la generación de datos digitales y cuantificables que pueden ser asignados al genoma. La SNG puede realizar un seguimiento de las alteraciones somáticas y hereditarias responsables por la expresión de RNAm aberrantes. Considerando la heterogeneidad de las lesiones orales, SNG puede ser ideal para estudiar y comprender las alteraciones genómicas que resultan en la formación de este grupo de tumores. Los hallazgos de los estudios con SNG de lesiones orales nos ayudarían a comprender mejor los aspectos genéticos de un tumor tradicionalmente considerado ambiental. Como un análisis sinérgico, se podrá mapear de nuevo los perfiles de RNAm alterados dentro de las regiones genómicas en que se encuentran mutaciones.
Las condiciones genéticas que han sido difíciles de diagnosticar debido a variantes raras o condiciones que tienen heterogeneidad genética sustancial, han estimulado la adopción de SNG en diferentes contextos clínicos59-60. En oncología, SNG se ha utilizado para la identificación de variantes raras y para lograr altas tasas de detección. Trujillano et al.61 evaluó recientemente el uso de un enfoque de enriquecimiento dirigido, combinado con la SNG multiplexada de secuenciación por síntesis en el análisis molecular de la fibrosis quística. Los autores informaron una tasa de detección de mutaciones de 100% y tasa de diagnóstico de 98,9%, y demostraron la capacidad de SNG para detectar grandes deleciones, duplicaciones e inversiones. Utilizando el analizador de DNA sistema MySeq se ha demostrado un rendimiento comparable, con una especificidad ≥ 99,99% en comparación con los métodos de referencia tradicionales. La alta exactitud de la SNG ofrece la promesa de detectar incluso variantes muy poco frecuentes para un gran número de enfermedades genéticas. Una vez que SNG es una clave para la obtención de objetivos diagnósticos, pronósticos y terapéuticos, estos marcadores deben ser validados en la evolución secuencial: Mutación – expresión de RNAm aberrante – cambios en niveles de proteínas61.
Se ha demostrado en varios tipos de tumores que el estado mutacional de los pacientes es clave en la determinación de la respuesta o en los niveles de respuesta al tratamiento62-63. El impulso de los ámbitos clínicos y de investigación para mejorar las tecnologías existentes ha dado lugar a la producción de una nueva generación de plataformas SNG con mayor precisión, eficacia en el tiempo y costo-efectivo. Se han desarrollado nuevos tipos de instrumentos de secuenciación que permiten una aceleración de las tasas de recopilación de datos para la secuenciación del DNA. La SNG ofrece la opción de producir más de mil millones de pares de bases en un trabajo de 4 días, pero se necesitan métodos sofisticados de bioinformática. El análisis y la traducción de estos datos en la clínica se complican aún más por la naturaleza heterogénea de la mayoría de los cánceres. Además, la mayoría de las muestras tumorales archivadas se conservan como tejido FFEP; Por lo tanto, la calidad del material de partida para estos estudios puede verse comprometida. A pesar de estas limitaciones, algunos estudios han utilizado con éxito muestras FFEP para estudiar mutaciones, variación del número de copias y determinación de variaciones de la línea germinal.64-66
Se espera que la SNG se expanda de una técnica enfocada en cáncer/mutación para incluir translocación, alteraciones del número de copias del gen y cambios epigenéticos del genoma. Con los avances en tecnología y nuestra comprensión sobre heterogeneidad intra-tumoral, la muestra inicial y el control de la línea germinal también pueden cambiar. Sin embargo, entre la producción de datos de SNG y la aplicación de estos hallazgos a las necesidades clínicas cotidianas, hay algunos obstáculos importantes a superar. En primer lugar, la detección de una alteración genética no necesariamente la convierte en una mutación iniciadora o driver. Un paso más cerca de la aplicación clínica, una mutación identificada no es necesariamente prevenible ni definitivamente tratable. Mutaciones de silenciamiento en los genes supresores de tumores son muy complejas a la inversa, y en muchos casos sus inhibidores no pueden ser usados67. La mayoría de las vías de señalización relacionadas con cáncer son muy críticas, y la inhibición o alteración de éstas podría ser letal; sin embargo, la presencia de vías de compensación hace que la letalidad sintética sea una estrategia exitosa68.
La heterogeneidad del carcinoma de células escamosas de cabeza y cuello, combinado con diferencias étnicas y geográficas en el genoma humano69, requiere una atención extrema a los detalles en el diseño de estudios con SNG. SNG tiene potencial para transformar el tratamiento de atención en salud oral, prediciendo activamente el riesgo y previniéndolos. Sin embargo, la optimización y estudios adicionales detallados son necesarios para arrojar luz sobre la patogénesis molecular del cáncer para ayudar a los pacientes en situaciones clínicas cotidianas de manera personalizada y significativa.
Por lo tanto, la nueva generación de clínicos de salud bucal debe reconocer que el término "cáncer de cabeza y cuello" no refleja adecuadamente la complejidad de este grupo de tumores de una manera como la defendida por Ellis y Perou69 en relación al cáncer de mama. En el cáncer de mama, desde principios del año 2000 ya se conocen por lo menos 5 grupos heterogéneos de patologías, todas definidas como cáncer de mama. Sin embargo, tanto el perfil mutacional como transcripcional difiere los subgrupos de estos tumores, La SNG es una herramienta potente, con costos cada vez más reducidos y presentes en muchos laboratorios de genética y biología molecular. El acceso de pacientes con patologías complejas incluyendo neoplasias malignas y benignas más que nunca pueden hacerse uso de esta tecnología. Por ejemplo, logramos conocer qué mutaciones iniciadoras están presentes en tejidos clínicamente benignos pero alterados y atribuir características genéticas que se asocien con el potencial de malignizarse. Como se ha señalado anteriormente, la aplicación de todas las tecnologías disponibles no hace que nuestra atención de pacientes sea personalizada, sino que nos ayuda a llegar a un "diagnóstico real" y una "etiología de concordancia para un tratamiento adecuado" que se convierte en atención estándar.70